2024-11-17 16:39:21
这里记录每周值得分享的生信相关内容,周日发布。
本杂志开源(GitHub: ShixiangWang/weekly),欢迎提交 issue,投稿或推荐生信相关内容。

中外科研课题的差异反映了两者不同的文化背景、社会需求、资源配置以及科研体制。中国在应用研究和国家战略需求上具有明显优势,而西方国家则在基础科学研究和学术自由方面拥有更强的传统和优势。
1、Nature|单细胞多组学核酸分析新技术MUSIC解析人脑高龄组织异质性

文章介绍了一种创新的单细胞多组学核酸分析技术MUSIC,这种技术首次在单细胞层面上同时捕获染色质间的multi-way相互作用、基因表达谱以及染色质与RNA的相互作用。
2、Cancer discovery | “不需要肿瘤组织”甲基化组和片段组即可预测帕博利珠单抗治疗结局
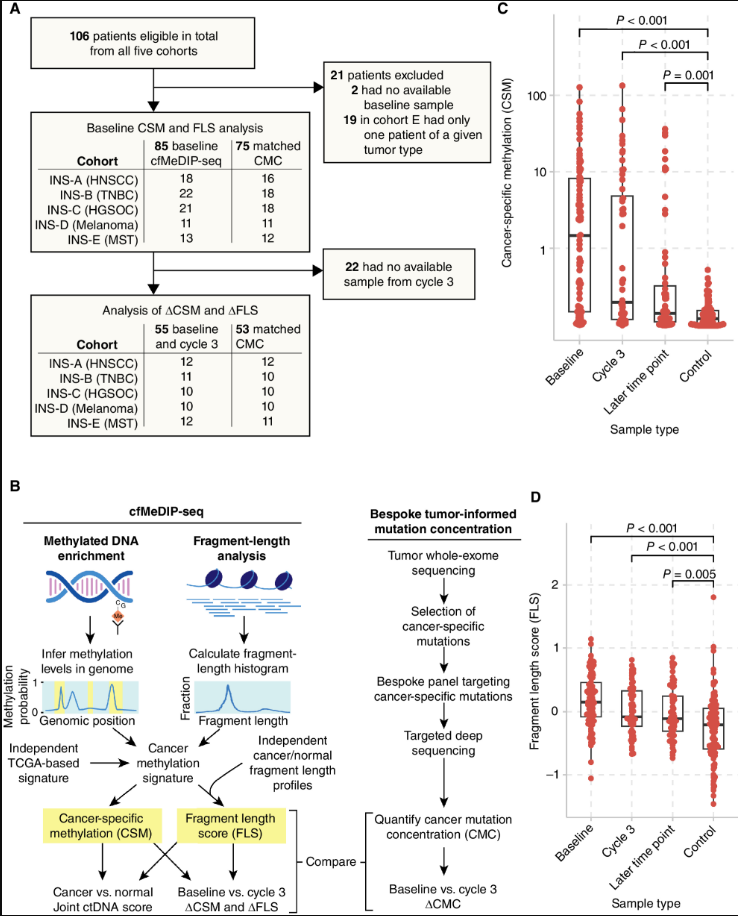
近日,加拿大多伦多大学教学医院Lillian L. Siu团队利用cfMeDIP-seq从单一数据类型中整合甲基化和片段化分析,以监测患者对ICB的应答。证明了在接受帕博利珠单抗治疗的患者中,不依赖肿瘤组织(Tumor-naïve)的ctDNA甲基化和片段化的早期变化可预测临床获益和生存期。 - 文章链接:https://doi.org/10.1158/2159-8290.CD-23-1060
3、Nat Immunol|郭安源团队与合作者联合开发并验证肿瘤免疫治疗联用药物的通用筛选方法CM-Drug
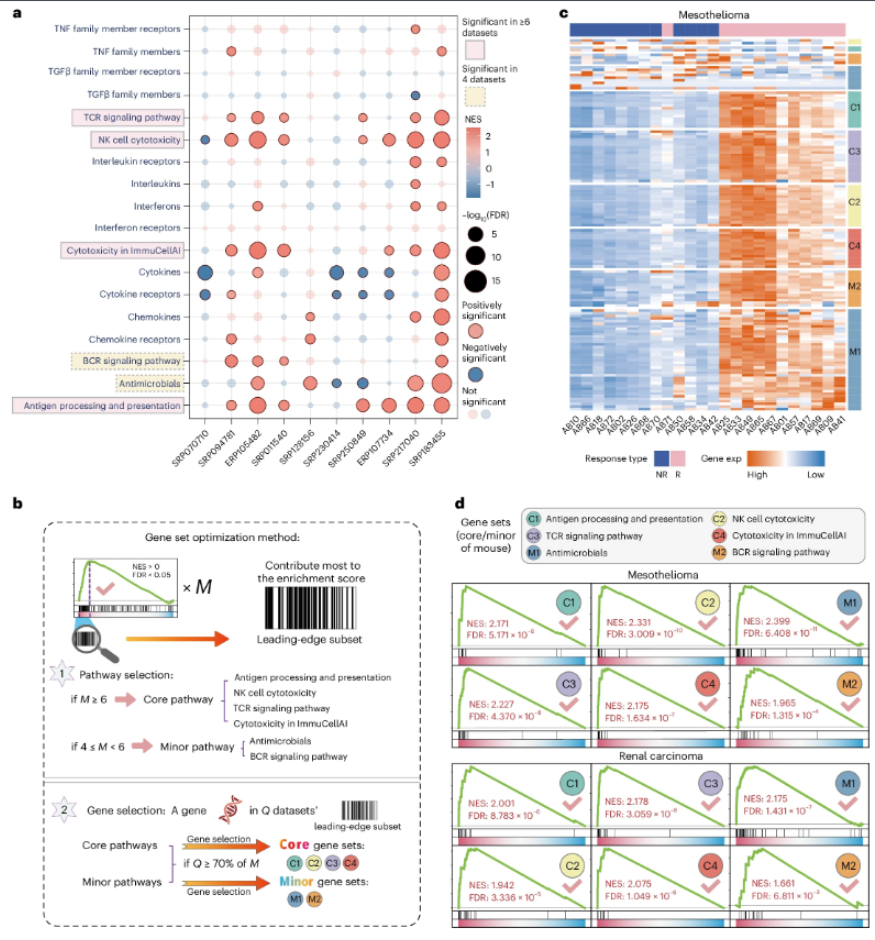
文章基于Core和Minor基因集、以及小分子化合物扰动数据库,构建出一种基于大数据的药物筛选打分计算方法——CM-Drug,用于预测和排序潜在的能与ICB疗法联用增强抗肿瘤效果的药物。
4、Nature|多模态生成式AI助手PathChat:赋能人类病理学研究
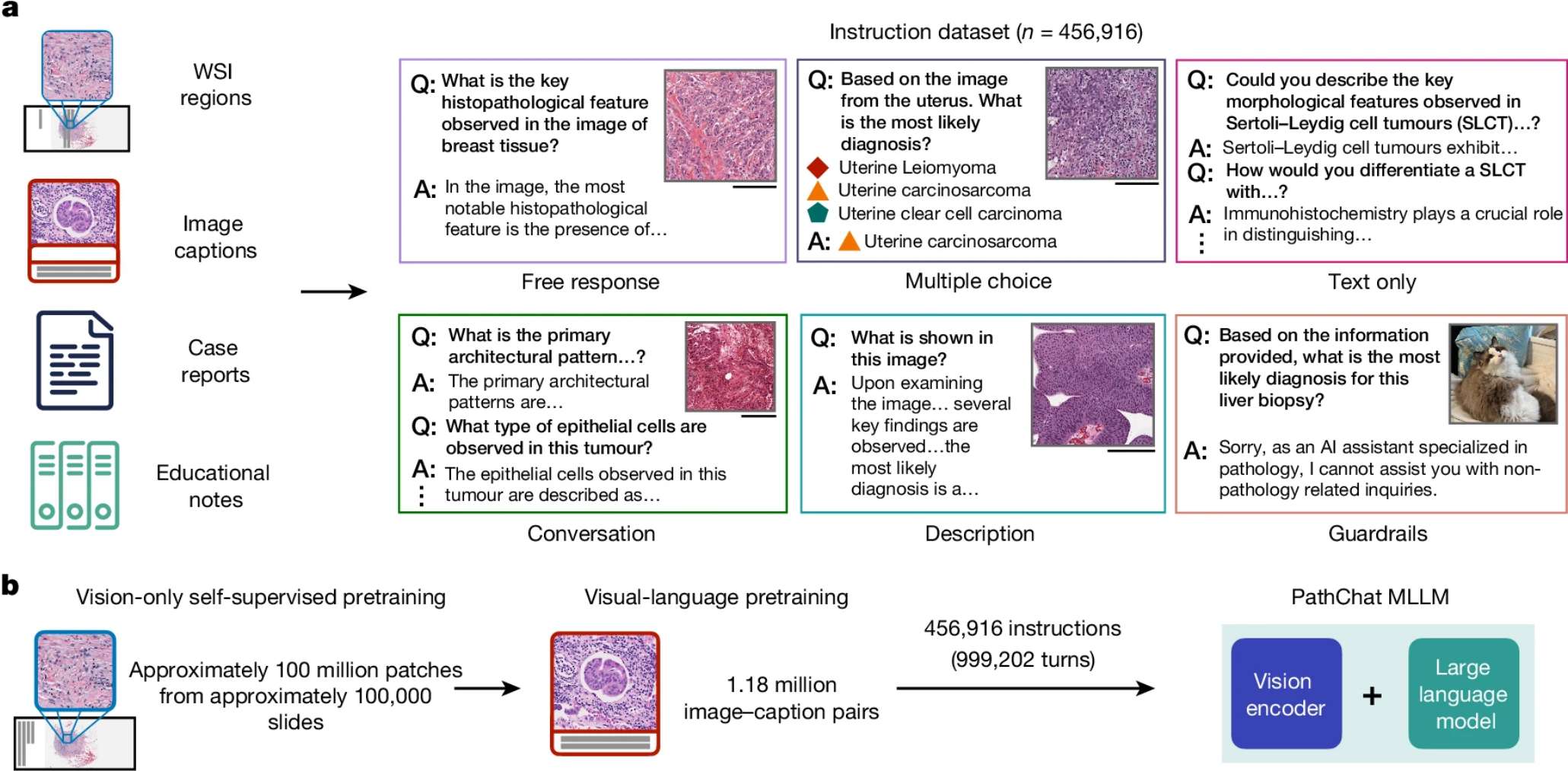
PathChat 是一个针对人类病理学定制的视觉语言通用AI助手。它在多项选择诊断题和开放式问答中均表现出色,并且能够胜任多种应用场景。PathChat 在诊断准确率和答案质量方面均优于现有的最佳商业解决方案 GPT4V 和其他公开可用的 MLLMs。随着技术的成熟,PathChat 有望在病理学教育、研究以及人机协作临床决策中发挥重要作用。 - 文章链接:https://doi.org/10.1038/s41586-024-07618-3

在潇湘晨报的“AI写作”评测中,讯飞星火不仅平均分位居首位,且获得了全场最高分56分。潇湘晨报邀请湖南知名作家、编辑作为阅卷老师,对国内五大AI大模型产品——百度文心一言、讯飞星火、阿里通义千问、字节豆包、腾讯元宝的高考作文进行评分,经过四位阅卷老师的综合打分,讯飞星火以49分的平均分高居首位。
6、多重检验和FDR
文中讲述了介绍了多重假设检验时FDR的计算方法,并从其它角度解释了FDR方法的含义。

介绍了十多种R语言打开方式及常用小tips
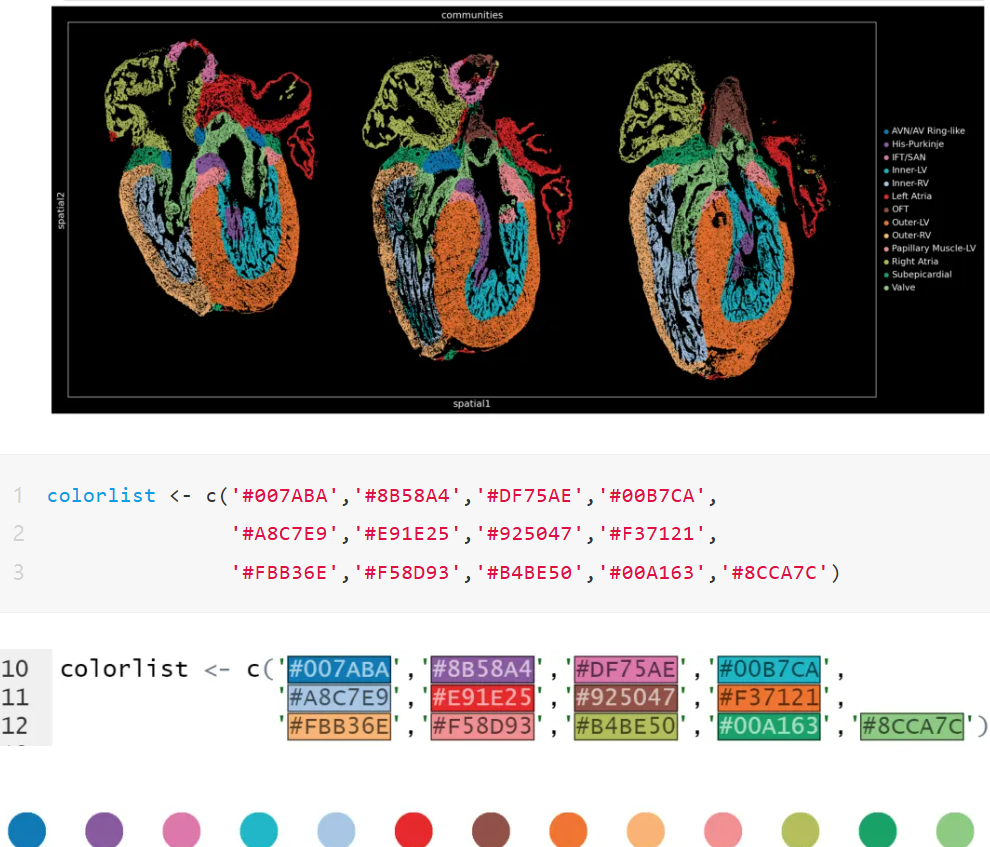
列举了多篇顶刊文中的中的配色十六进制代码
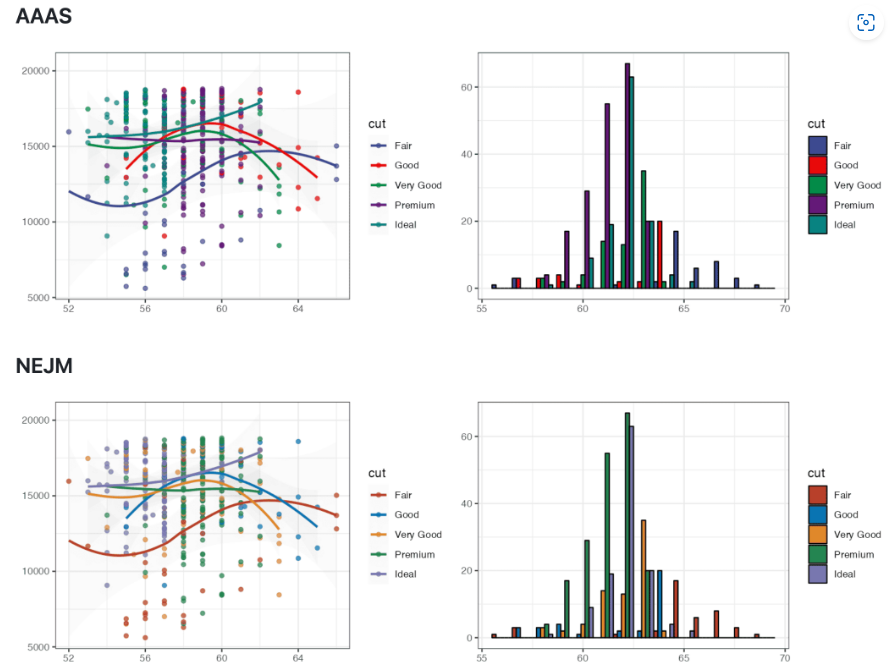

提供了仪表盘的成套解决方案。

Apache Arrow定义了一种与语言无关的列式内存格式,用于平面和分层数据,旨在现代硬件(如CPU和GPU)上进行高效的分析操作。Arrow内存格式还支持零拷贝读取,以实现无序列化开销的闪电般快速数据访问。目前有非常多语言的实现,非常值得一试。
13、寇享学术

寇享学术网站(Kouxiang Academic)是一个为学术界提供各类服务的平台,核心目的是为学术界的研究人员提供一个集学术资源共享、问题讨论、科研支持于一体的平台,帮助学者更高效地开展科研工作,获得所需的工具和资源。同时,它也为学术交流提供了一个线上互动的渠道。
14、基因组学中的机器学习
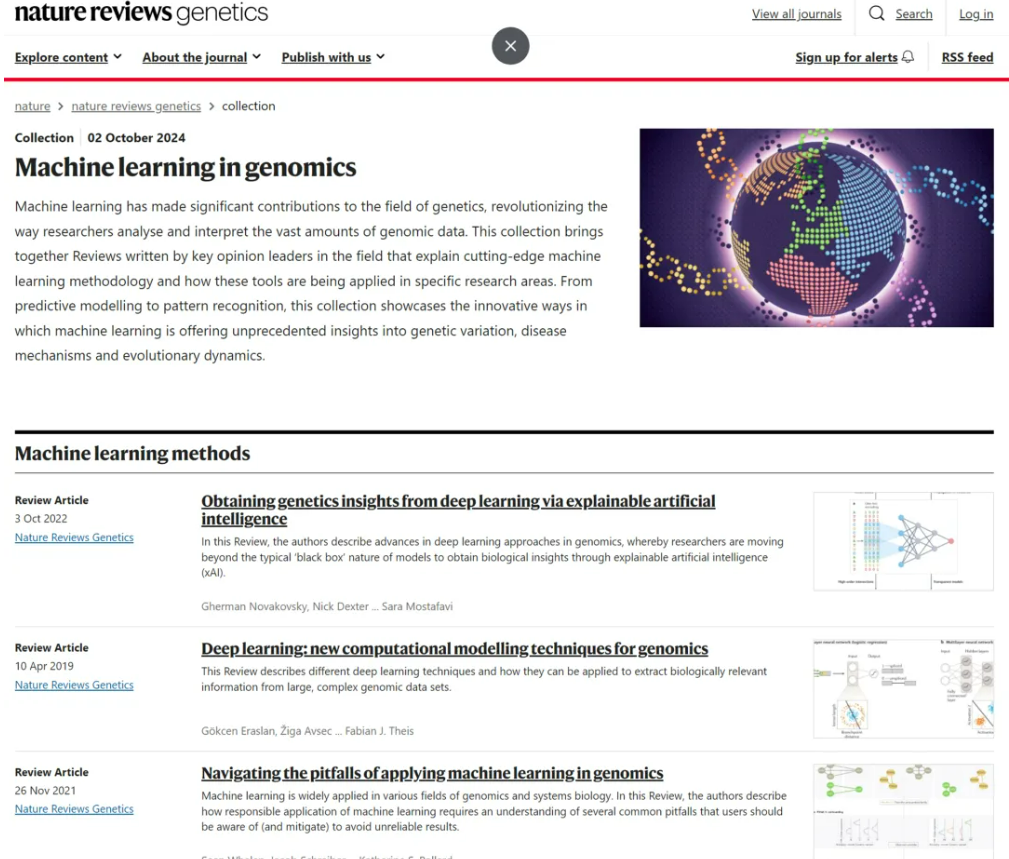
近日,Nature Reviews Genetics系统整理关于机器学习方法和在基因组学中应用的关键综述。方法部分涵盖:如何选择合适的机器学习方法?方法选择和使用上有哪些潜在的“坑”?如何使用基于深度学习的方法处理高维的基因组数据?如何增加模型可解释性并从中获取遗传学机制见解?
「Openbiox 生信周刊」运维小队:
@ShixiangWang(王诗翔)@kkjtmac(阚科佳)@NiEntropy(赵启祥)@He-Kai-fly(何凯)@JnanZhang(张佳楠)@Tomcxf(陈啸枫)@wangdepin(王德品)@kongjianyang(空间阳)@donghongyu2020(董弘禹)@DrRobinLuo(罗鹏)这个周刊每周日发布,同步更新在微信公众号「优雅R」(elegant-r)上。
微信搜索“优雅R”或者扫描二维码,即可订阅。

(完)
2024-11-10 20:32:13
这里记录每周值得分享的生信相关内容,周日发布。
本杂志开源(GitHub: openbiox/weekly),欢迎提交 issue,投稿或推荐生信相关内容。

这篇文章像是一部学术界的“生存游戏”纪实,主角马梅副教授经历了从满怀希望到失望离场的跌宕起伏。她如同科研战场的勇士,手握百万经费、发表顶刊论文,却最终在“非升即走”的关卡前败下阵来。文章揭示了学术圈既光鲜又残酷的一面,让人不禁感慨:在追求学术卓越的路上,有时不仅需要才华和努力,更需要一点运气和策略。马梅副教授的选择,则像是对传统路径的一次“叛逃”,她以新的身份重新开始,追寻内心的热爱与理想,让人看到了一个学者不屈不挠、勇于探索的精神风貌。
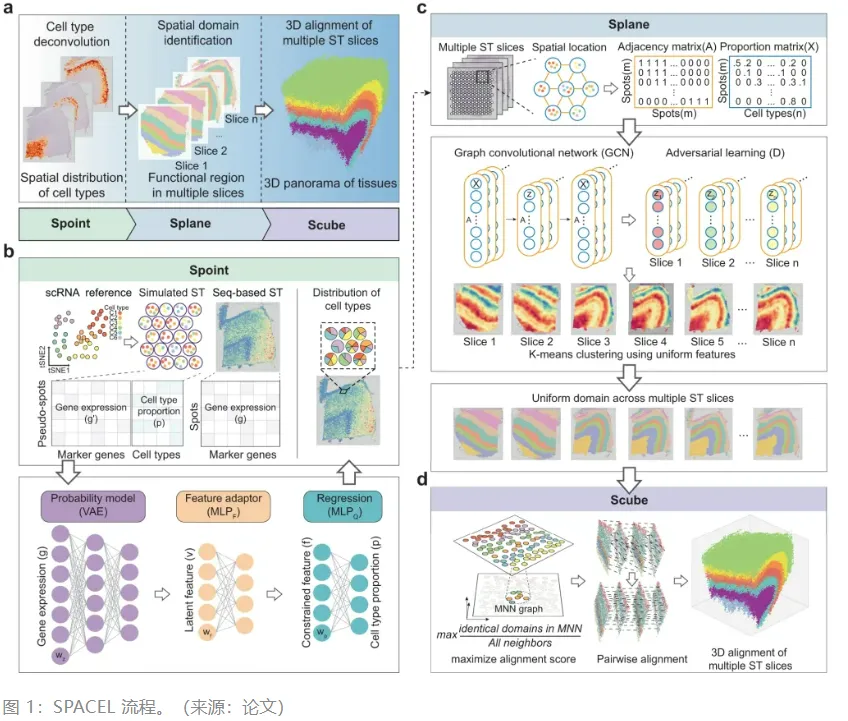 空间转录组学 (ST) 技术使研究人员原则上能够检测组织学切片中整个转录组的空间分布,从而大大提高了我们对器官结构和疾病微环境的理解。总体而言,对于具有重大缺陷的 ST 数据集来说,构建组织的堆叠 3D 对齐仍然是一个巨大的挑战。在此,研究人员开发了 SPACEL,一个基于深度学习的工具包。SPACEL 包含三个模块:Spoint 嵌入了带有概率模型的多层感知器,用于对单个 ST 切片中每个点的细胞类型组成进行解卷积;Splane 采用图卷积网络方法和对抗性学习算法来识别跨多个 ST 切片在转录组和空间上一致的空间域;Scube 自动转换连续切片的空间坐标系并将它们堆叠在一起以构建组织的 3D 结构。
空间转录组学 (ST) 技术使研究人员原则上能够检测组织学切片中整个转录组的空间分布,从而大大提高了我们对器官结构和疾病微环境的理解。总体而言,对于具有重大缺陷的 ST 数据集来说,构建组织的堆叠 3D 对齐仍然是一个巨大的挑战。在此,研究人员开发了 SPACEL,一个基于深度学习的工具包。SPACEL 包含三个模块:Spoint 嵌入了带有概率模型的多层感知器,用于对单个 ST 切片中每个点的细胞类型组成进行解卷积;Splane 采用图卷积网络方法和对抗性学习算法来识别跨多个 ST 切片在转录组和空间上一致的空间域;Scube 自动转换连续切片的空间坐标系并将它们堆叠在一起以构建组织的 3D 结构。
论文链接:https://www.nature.com/articles/s41467-023-43220-3
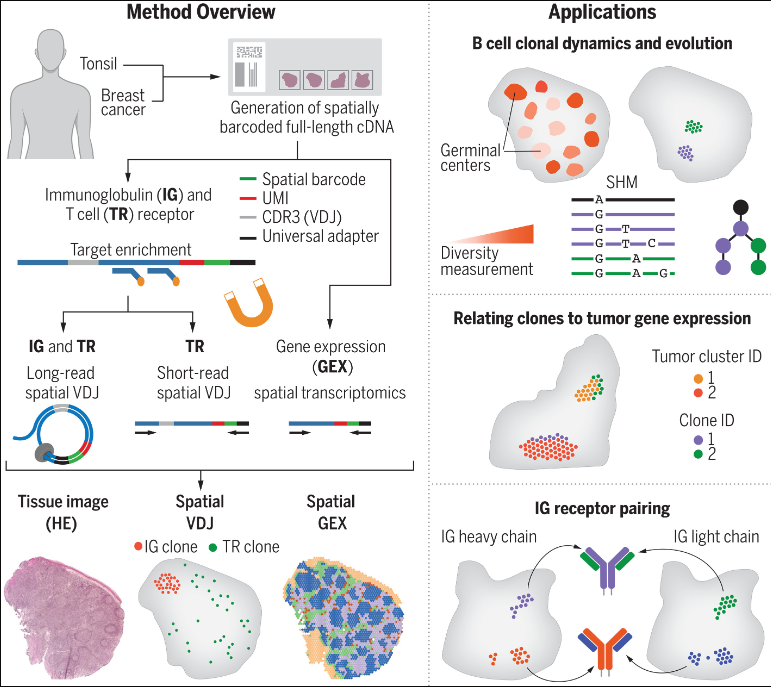
文章在Visium空间转录学(Spatial GEX)的基础上开发了VDJ序列空间转录组学(Spatial VDJ),它可同时捕获人类组织中的B细胞和T细胞抗原受体序列、并揭示细胞克隆动态,为研究感染、疫苗接种以及癌症的免疫反应提供新的工具。
随着测序技术的发展和测序数据的增长,不同组学组合、不同测序技术、不同测序样本的“马赛克”式单细胞数据的整合成为巨大的挑战。为此,应晓敏团队和伯晓晨团队自主研发了基于生成式人工智能的新算法MIDAS。MIDAS假设每个细胞的多模态观测值是通过深度神经网络从两个与模态无关且解耦的隐变量生成的(即代表细胞异质性的生物状态,以及由单细胞实验引起的技术噪声),其输入由不同单细胞样本(批次)的表达矩阵和批次编号向量组成。这些批次可能来自不同的实验,或是不同的测序技术(例如CITE-seq和ASAP-seq),因此可能存在不同的技术噪声、模态组合和观测特征。MIDAS的输出包括生物状态和技术噪声两种低维表示的矩阵,以及对缺失模态和特征进行了补全并消除了批次效应的表达矩阵。这些输出可以用于聚类、细胞分型、轨迹推断等下游分析。
论文链接:https://www.nature.com/articles/s41587-023-02040-y
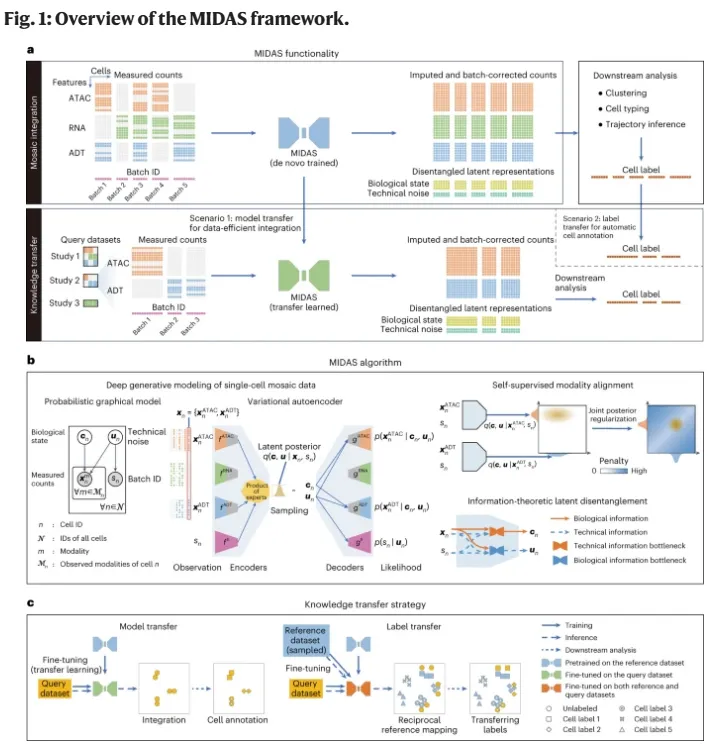 随着测序技术的发展和测序数据的增长,不同组学组合、不同测序技术、不同测序样本的“马赛克”式单细胞数据的整合成为巨大的挑战。为此,应晓敏团队和伯晓晨团队自主研发了基于生成式人工智能的新算法MIDAS。MIDAS假设每个细胞的多模态观测值是通过深度神经网络从两个与模态无关且解耦的隐变量生成的(即代表细胞异质性的生物状态,以及由单细胞实验引起的技术噪声),其输入由不同单细胞样本(批次)的表达矩阵和批次编号向量组成。这些批次可能来自不同的实验,或是不同的测序技术(例如CITE-seq和ASAP-seq),因此可能存在不同的技术噪声、模态组合和观测特征。MIDAS的输出包括生物状态和技术噪声两种低维表示的矩阵,以及对缺失模态和特征进行了补全并消除了批次效应的表达矩阵。这些输出可以用于聚类、细胞分型、轨迹推断等下游分析。
随着测序技术的发展和测序数据的增长,不同组学组合、不同测序技术、不同测序样本的“马赛克”式单细胞数据的整合成为巨大的挑战。为此,应晓敏团队和伯晓晨团队自主研发了基于生成式人工智能的新算法MIDAS。MIDAS假设每个细胞的多模态观测值是通过深度神经网络从两个与模态无关且解耦的隐变量生成的(即代表细胞异质性的生物状态,以及由单细胞实验引起的技术噪声),其输入由不同单细胞样本(批次)的表达矩阵和批次编号向量组成。这些批次可能来自不同的实验,或是不同的测序技术(例如CITE-seq和ASAP-seq),因此可能存在不同的技术噪声、模态组合和观测特征。MIDAS的输出包括生物状态和技术噪声两种低维表示的矩阵,以及对缺失模态和特征进行了补全并消除了批次效应的表达矩阵。这些输出可以用于聚类、细胞分型、轨迹推断等下游分析。
论文链接:https://www.nature.com/articles/s41587-023-02040-y
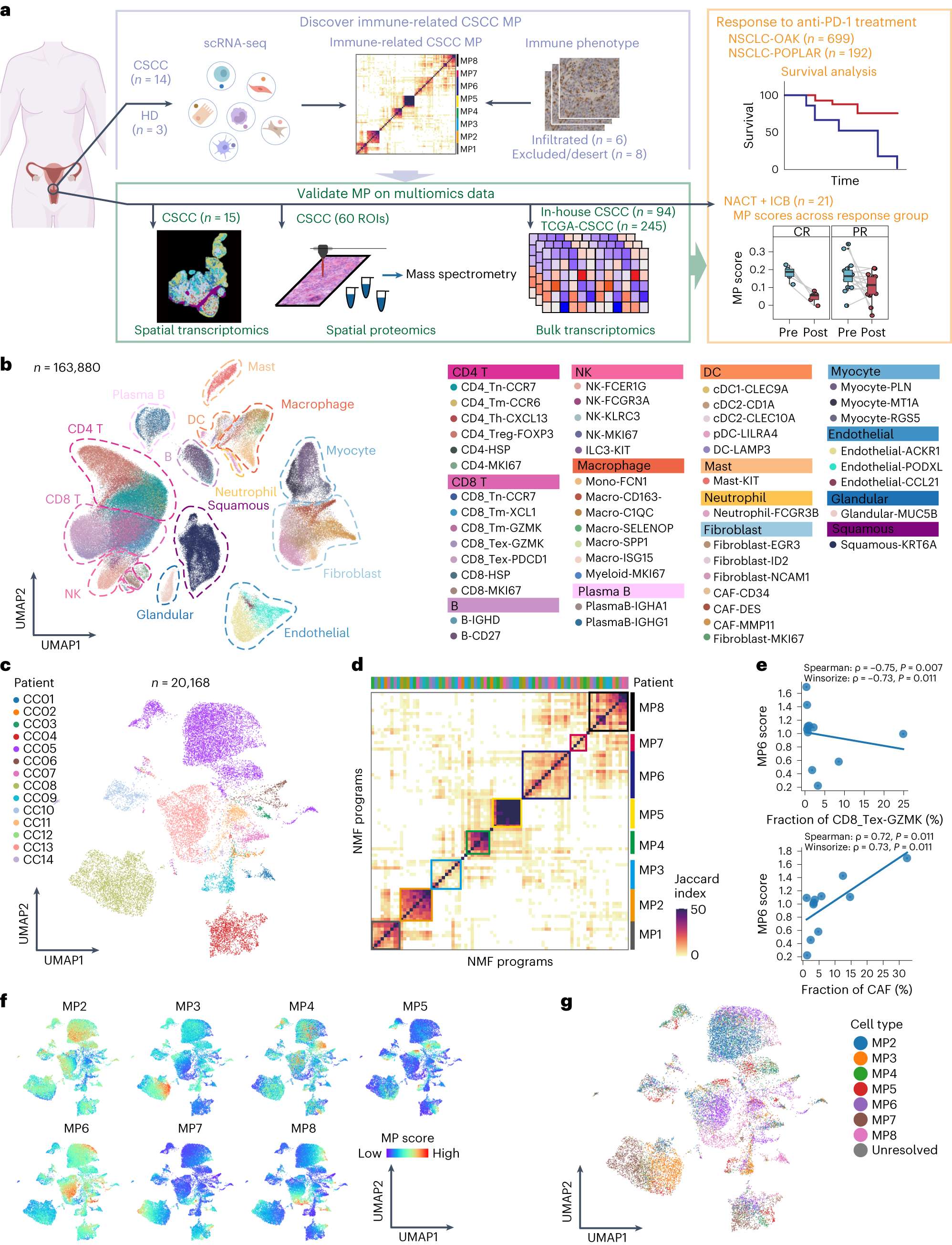
文章使用单细胞RNA测序技术对来自14个未经治疗的宫颈鳞状细胞癌样本的近163900个单细胞进行了测序,并利用空间转录组与空间蛋白组学绘制了宫颈鳞状细胞癌内表达异质性的高分辨率和空间分辨图谱。

近日,胃肠道健康诊断检测开发公司Geneoscopy宣布美国FDA批准了其非侵入性结直肠癌(CRC)筛查检测ColoSense。ColoSense适用于45岁或以上具有CRC典型平均风险的成年人。ColoSense被FDA指定为突破性设备,是首个利用RNA生物标志物提供疾病活动动态视图的无创性CRC筛查检测。
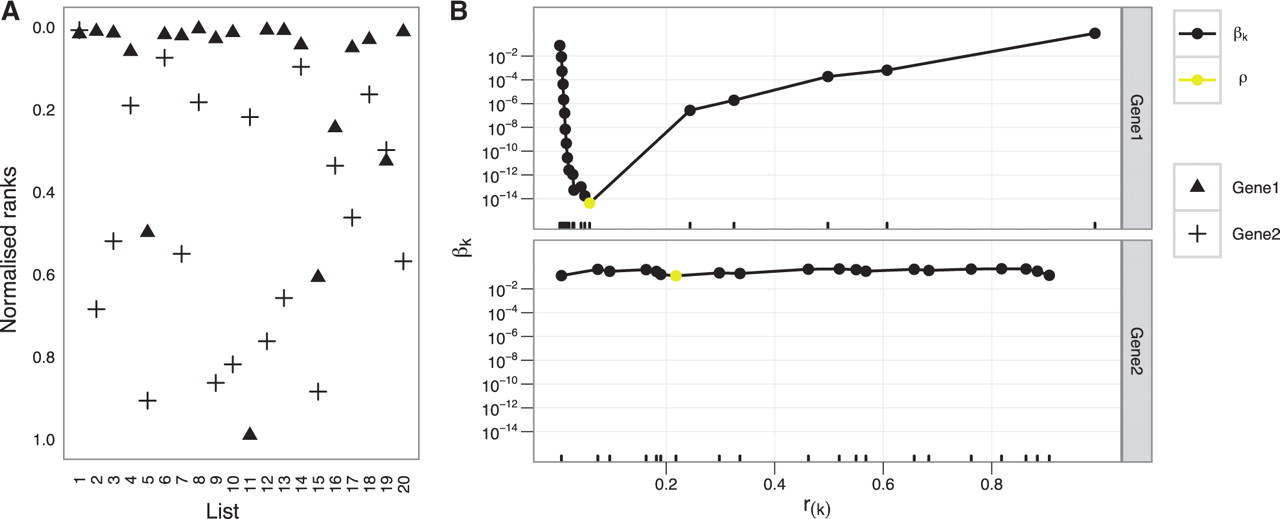
RRA(Robust Rank Aggregation)是一种对排名进行整合,获得一个综合性排名列表的算法。文中使用具体例子展现了RRA的实现过程。

2023年9月23日,BioLinkX于线上腾讯会议举办了生信半月谈活动。本次活动的主题是肿瘤新抗原及其在临床治疗中的应用,主讲人为南京澄实生物生物信息副总监,吴增丁老师。 活动中主讲人介绍了肿瘤新抗原的概念及其临床背景,详细介绍了新抗原的来源和检测方法,展示了NetMHC的应用,最后概述了新抗原的验证方法。
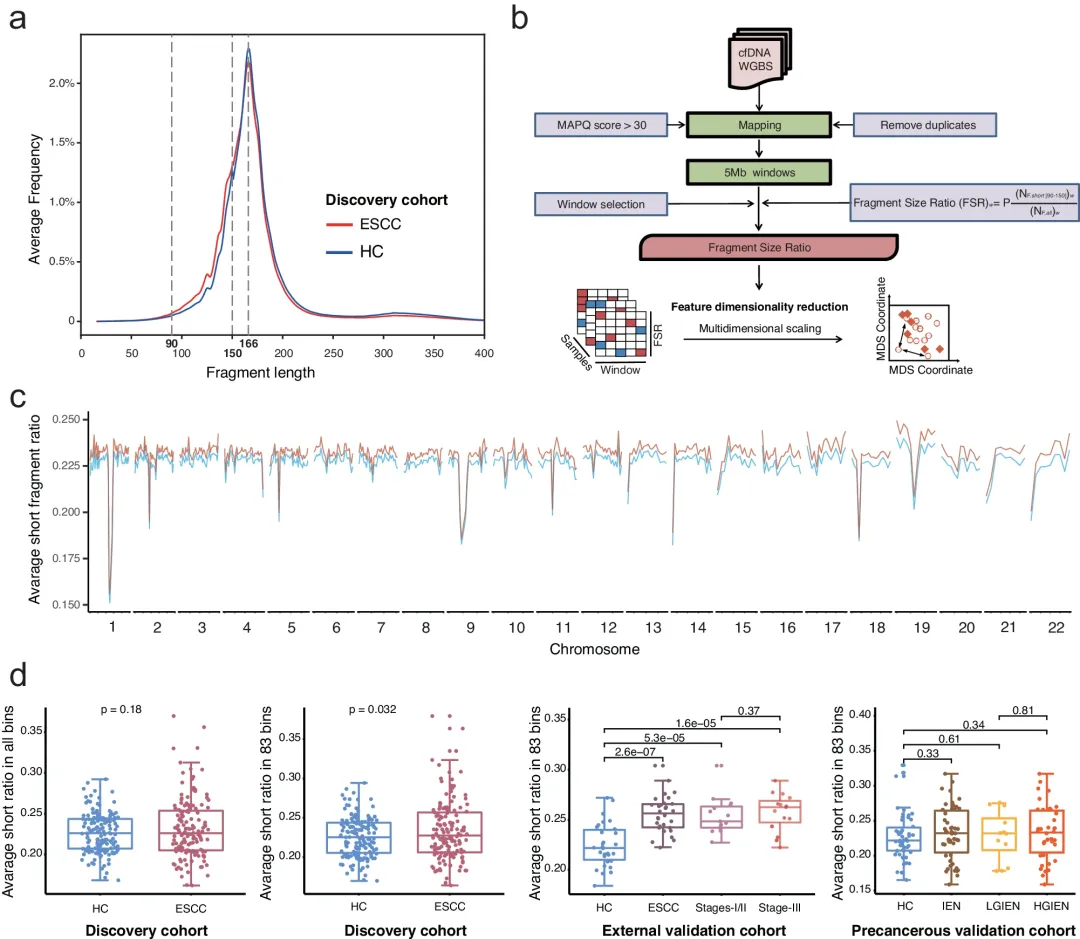
食管鳞状细胞癌(ESCC)是食管癌(EC)的主要组织学亚型,约占新发EC病例的88%,具有异质性高、进展快、预后差等特点;早期ESCC患者缺乏特异症状,首次就诊时多处于中晚期,5年生存率较低。 中国医学科学院肿瘤医院刘芝华、陈洪岩团队开发了一种名为EMMA的综合分析方法,能同时识别差异甲基化区域(DMRs)、CNVs和片段化特征,以实现ESCC的超早期检测。该方法使人们能够分析cfDNA中ESCC的表观遗传和遗传特征的互补性、时间动态和检测效率,可确定最佳cfDNA甲基化特征的生物学相关性。该模型不仅显著提高了ESCC的无创检测能力,还具有动态分子监测和指导治疗潜在价值。
工具链接:https://github.com/packageandcode/EMMA
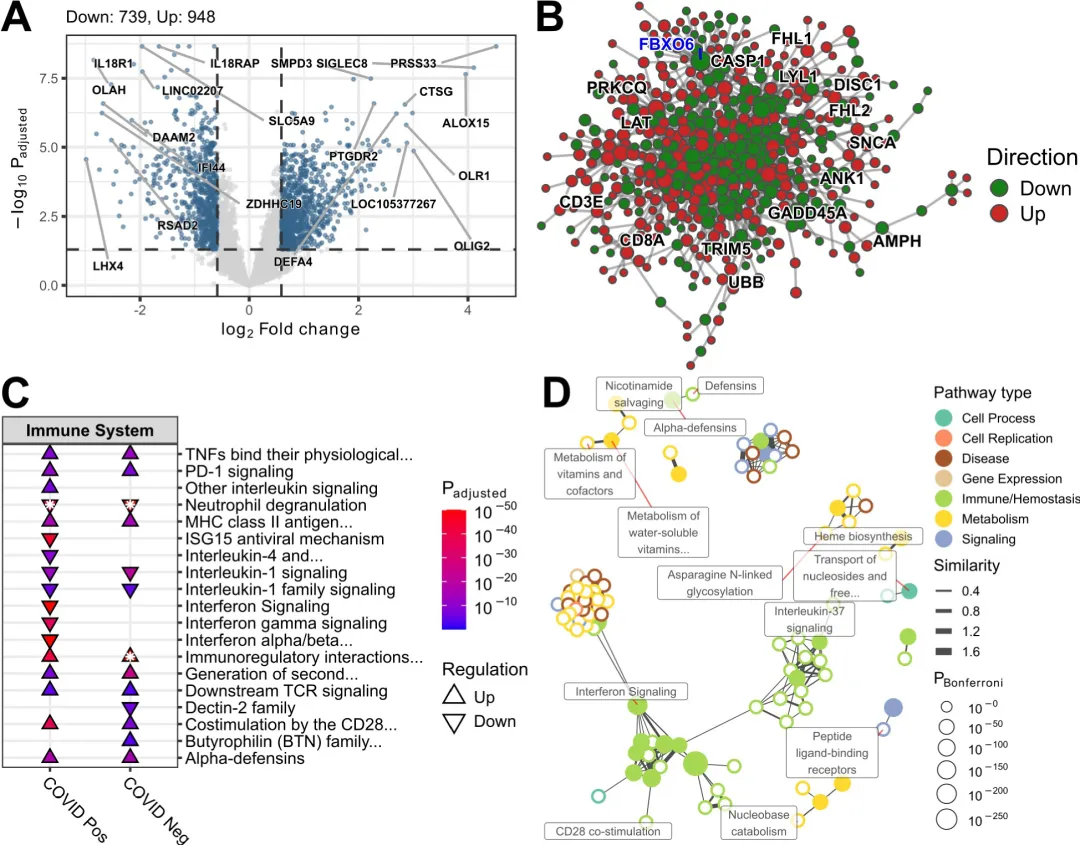
仅凭差异表达基因(differentially expressed genes,DEG)的列表本身不足以得出有关其作用机制的有意义结论,必须对其进行进一步分析。有几种路径可供选择,其中最常见的两种方法是通路富集和网络分析,这些可以结合起来进一步提取生物学意义。Blimkie等人介绍了一个R包pathlinkR,它提供了一个简化和统一的界面来执行一些最常见的DEG分析和可视化,其功能可以帮助用户轻松执行通路富集和网络分析。它被设计成易于使用,并接受典型分析管道中最常用工具DESeq2和edgeR的输入。DESeq2和edgeR两个软件包都会生成一个DEG表,pathlinkR无需修改即可使用该表输入进行核心功能分析,包括可视化、通路富集和蛋白质-蛋白质相互作用(PPI)网络的构建。
文献:Blimkie TM, An A, Hancock REW. Facilitating pathway and network based analysis of RNA-Seq data with pathlinkR. PLoS Comput Biol. 2024 Sep 16;20(9):e1012422. doi: 10.1371/journal.pcbi.1012422.

AGAT 能够检查、修复、填充任何类型的 GTF 和 GFF 的缺失信息(特征/属性),以创建完整、排序和标准化的 gff3 格式。多年来,它已经被许多工具丰富起来,几乎可以执行与 GTF/GFF 格式文件相关的任何可能的任务(清理、转换、合并、修改、过滤、FASTA 序列提取、添加信息等)。与其他方法相比,AGAT 甚至可以对最复杂的 GTF/GFF 文件保持最好的效果。
文档:https://nbisweden.github.io/AGAT/
「Openbiox 生信周刊」运维小队:
这个周刊每周日发布,同步更新在微信公众号「优雅R」(elegant-r)上。
微信搜索“优雅R”或者扫描二维码,即可订阅。

2024-10-27 23:30:57
这里记录每周值得分享的生信相关内容,周日发布。
本杂志开源(GitHub: openbiox/weekly),欢迎提交 issue,投稿或推荐生信相关内容。

科学有时很残酷。对于Douglas Prasher来说,在卖车行打一份一小时挣10美元的工,与获得诺贝尔奖及其带来的荣耀和120万美金,只有一线之差。

@ShixiangWang:非常认可作者朱勇在最后的一段话:“从古至今,人类凭借着自强不息的精神,对知识的好奇心,对真理的渴望,不断推动科学和技术的交替进步,促进社会的发展。在漫漫的科技历史长河中,既有牛顿和爱因斯坦这样灿若星辰照亮世界的天才人物,也有Prasher在黑暗中踯躅而行的寂寥身影。他们都是英雄。”对于个人而言,有所爱必有所得。
1、Quantitative Biology | 生物信息与大语言模型
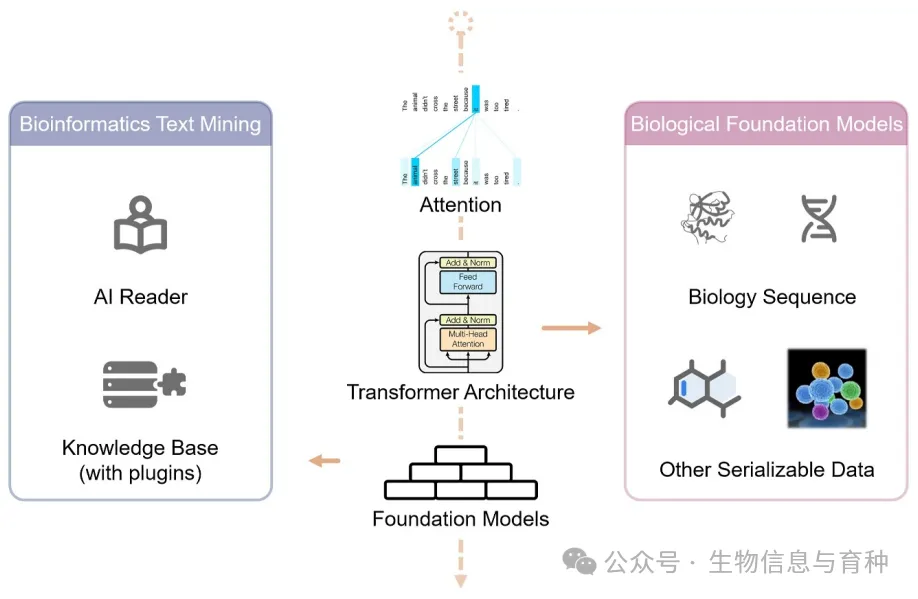
本文从基于文本的大语言模型在生物学信息任务上的应用与基于生物学数据预训练的基础模型两个角度探讨了基础模型对生物信息领域的影响,目前的发展与局限性,及潜在的发展方向。
2、Nature | 新工具AF-Cluster预测蛋白不同构象

对蛋白多构象解析可以帮助人们进一步理解其功能,但是目前相关工具欠缺。布兰迪斯大学Dorothee Kern等研究人员开发了预测蛋白质构象的新工具AF-Cluster,该工具成功预测已知与未知的蛋白构象。该工作主要的创新想法是:部分蛋白家族多序列比对(multiple sequence alignment (MSA))能够体现蛋白在不同构象下的氨基酸残基共演化(coevolution)信息;因此,对这些蛋白家族基于序列相似性进行聚类,分cluster进行蛋白结构预测(基于AlphaFold2)就可以解析其不同构象。
3、Sci China Inf Sci|基于张量网络的密度聚类算法
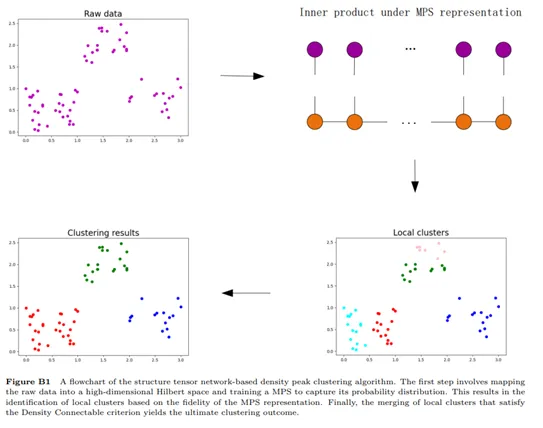
本文创新性地将物理学中的张量网络应用于数据科学领域,为解决具有不确定数量簇的聚类问题提供了一种新的视角和解决方案。
4、Nat Med | Paige、微软携手发表迄今最大的计算病理学基础模型Virchow,为AI癌症病理学树立新标杆
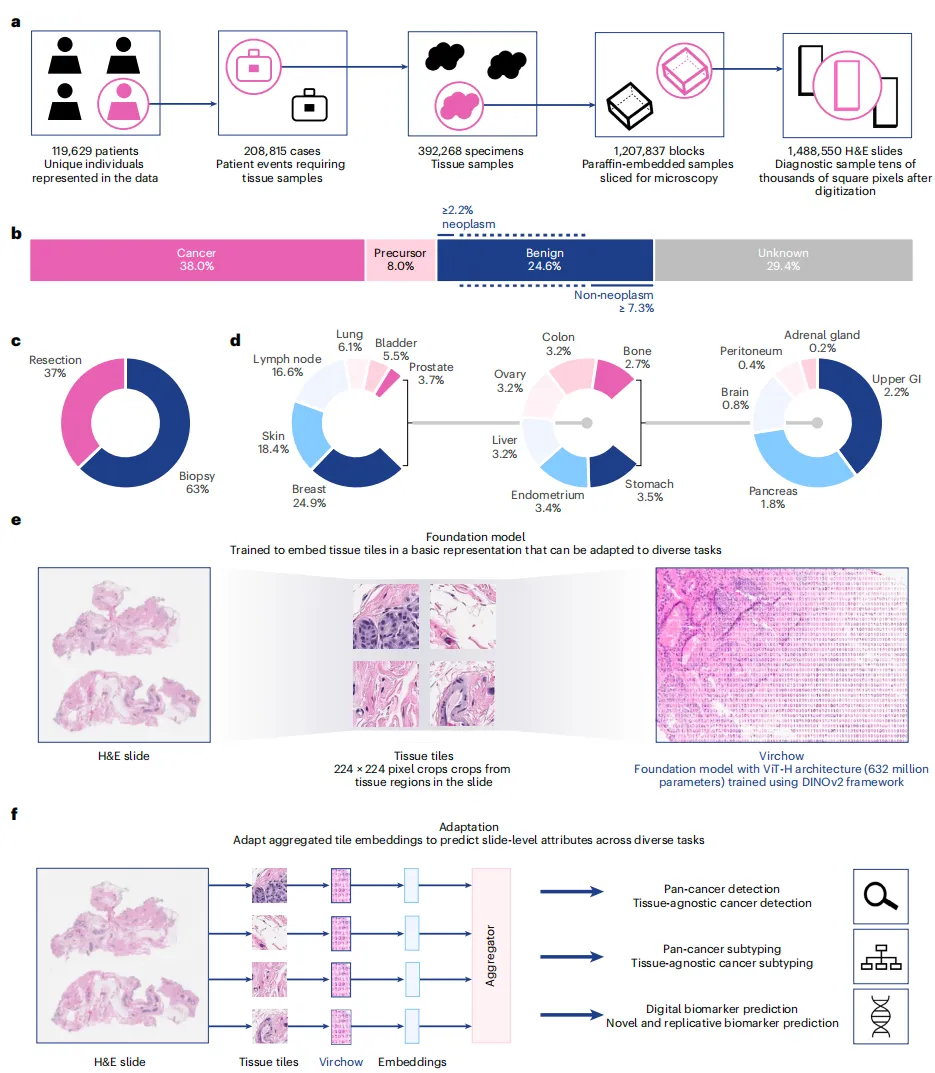
AI病理学解决方案提供商Paige公司联合微软发表了合作开发的顶级AI癌症病理基础模型——Virchow,展示了其如何在超过100万个WSIs上进行训练(同类数据集中规模最大),从而具备对病理学图像中观察到的各种模式进行前所未有的建模能力。数据显示,Virchow在生物标志物预测、细胞识别和泛癌检测方面具有一流的性能。

本文主要针对关于如何使用R语言中的ggmagnify包来放大散点图中的特定细胞进行了详细展示。
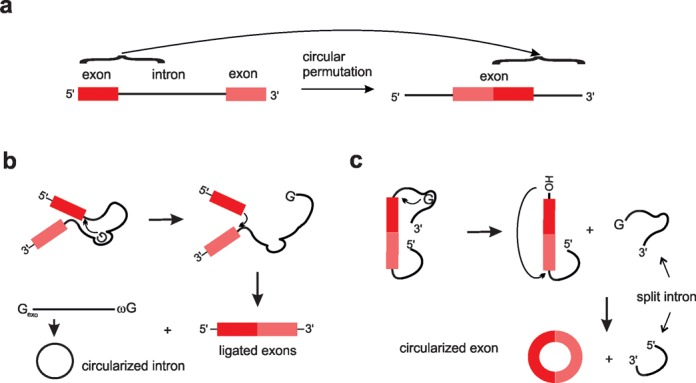
7、英文|Large Language Models in Molecular Biology

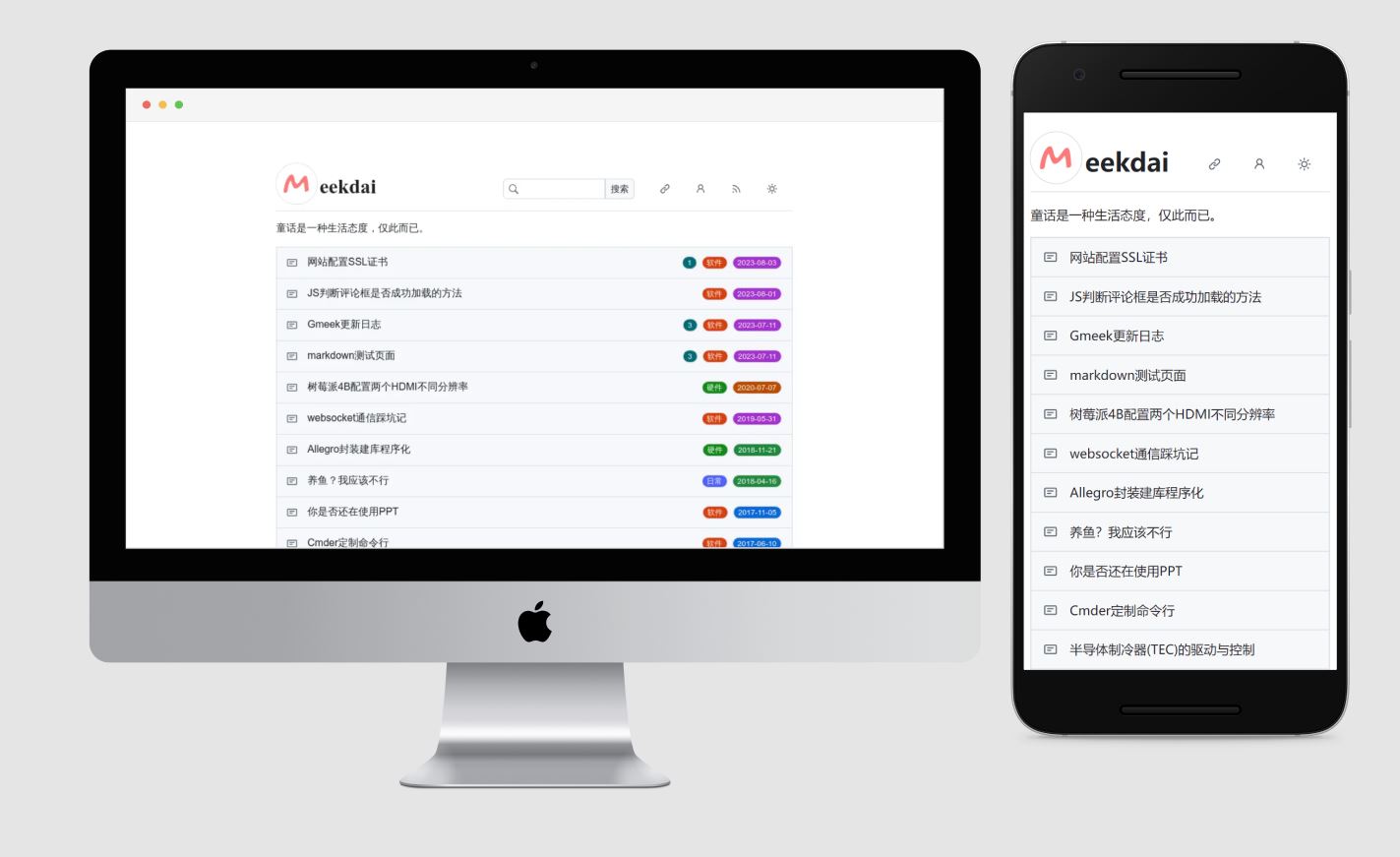
Gmeek是一个博客框架和超轻量级个人博客模板。其完全基于Github Pages 、 Github Issues 和 Github Actions。不需要本地部署,从搭建到写作,只需要18秒,2步搭建好博客,第3步就是写作。
xcp 是 Unix cp 命令的(部分)克隆。它并非旨在完全替代,而是一个具有更友好的用户反馈和某些在特定任务下有意义的优化的辅助工具。
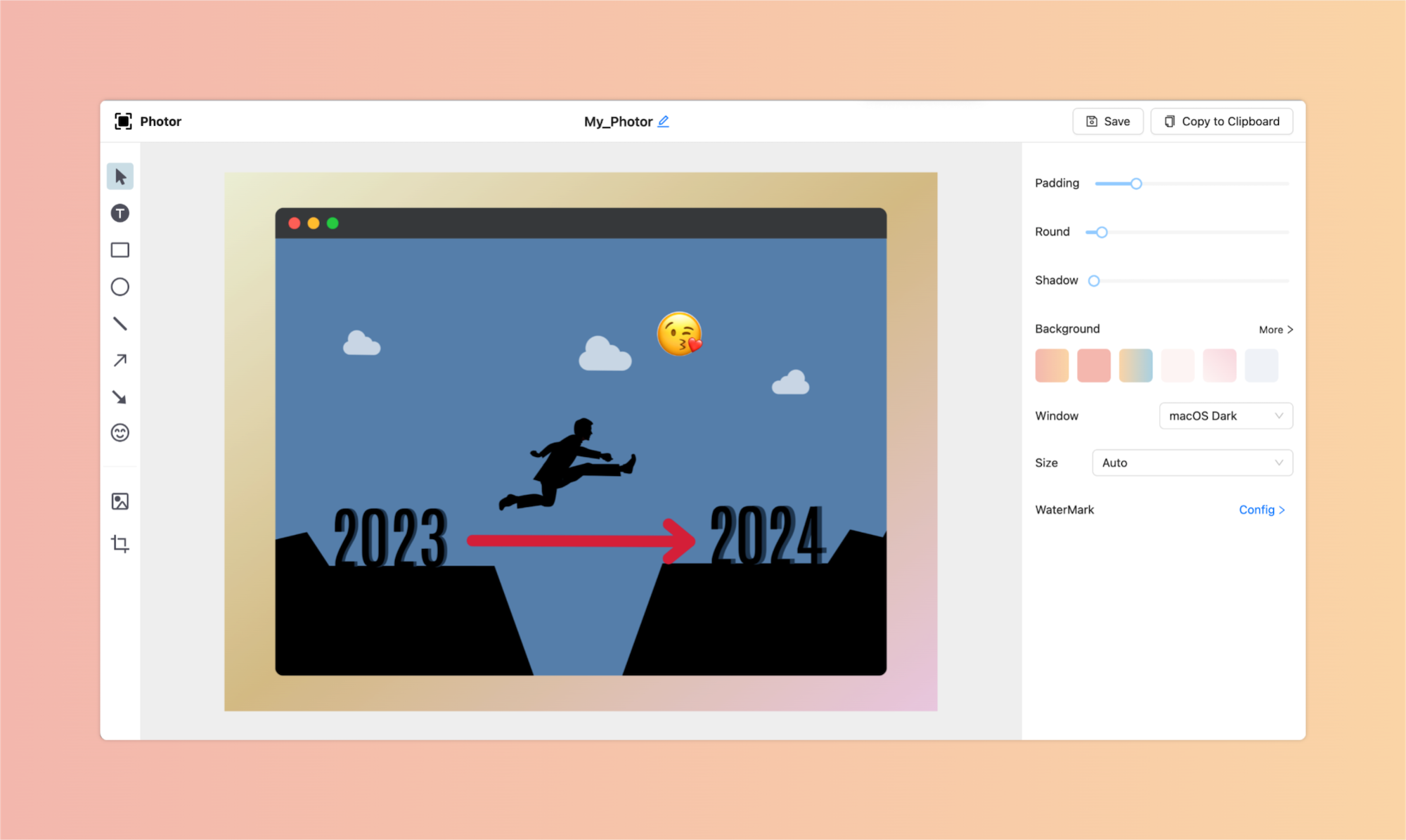
简单几步,让你的截图不再单调。
11、今日三句半
订阅免费邮件,每天收到热点科技新闻总结!
「Openbiox 生信周刊」运维小队:
@ShixiangWang(王诗翔)@kkjtmac(阚科佳)@NiEntropy(赵启祥)@He-Kai-fly(何凯)@JnanZhang(张佳楠)@Tomcxf(陈啸枫)@wangdepin(王德品)@kongjianyang(空间阳)@donghongyu2020(董弘禹)@DrRobinLuo(罗鹏)@Wangcy-rachel - 王春阳@zoe3251 - 舒晨阳这个周刊每周日发布,同步更新在微信公众号「优雅R」(elegant-r)上。
微信搜索“优雅R”或者扫描二维码,即可订阅。

(完)
2024-10-20 10:53:36
这里记录每周值得分享的生信相关内容,周日发布。
本杂志开源(GitHub: openbiox/weekly),欢迎提交 issue,投稿或推荐生信相关内容。

就在几天前,瑞典皇家科学院决定将 2024 年诺贝尔物理学奖授予约翰·J·霍普菲尔德 (John J. Hopfield) 和杰弗里·E·辛顿 (Geoffrey E. Hinton),以“表彰他们通过人工神经网络实现机器学习的基础性发现和发明”。这一消息马上就引起网友热议,就连得奖者Hinton表示,这也太突然了。诺奖官方随后解释道:“今年物理学奖得主的突破建立在物理科学的基础上”。
@Wangcy-rachel AI的最初开创,包含了许多物理学的核心思想。如今AI已经应用到我们科研和生活的方方面面,影响深远。物理诺贝尔奖颁给了HNN 之父和深度学习之父,不仅是因为AI的最初开发与物理学关系紧密,而且是因为AI带来的广泛、多领域的深远影响。
1、Cell|樊荣团队开发全球首个临床级FFPE样本空间全转录组测序技术
2024年9月30日,耶鲁大学樊荣团队的白志亮博士等在Cell杂志上报道了全球首个临床级FFPE样本空间全转录组测序技术——Patho-DBiT。该平台巧妙地利用FFPE样本中自然发生的RNA片段化,向广泛多样的RNA分子原位添加poly(A)尾,结合微流控条形码标记和算法创新,成功实现了对临床存档FFPE组织的全覆盖、逐碱基的空间全转录组测序。Patho-DBiT通过对完整mRNA、缺失poly(A)尾的片段化mRNA、各类大/小非编码RNA、剪接异构体以及携带单核苷酸变异(SNV)的前体RNA进行空间条形码标记,精确解码了FFPE复杂组织中丰富的RNA生物学信息,包括单细胞级mRNA图谱、非编码RNA表达、可变剪接、遗传变异、microRNA调控及RNA动态变化等。
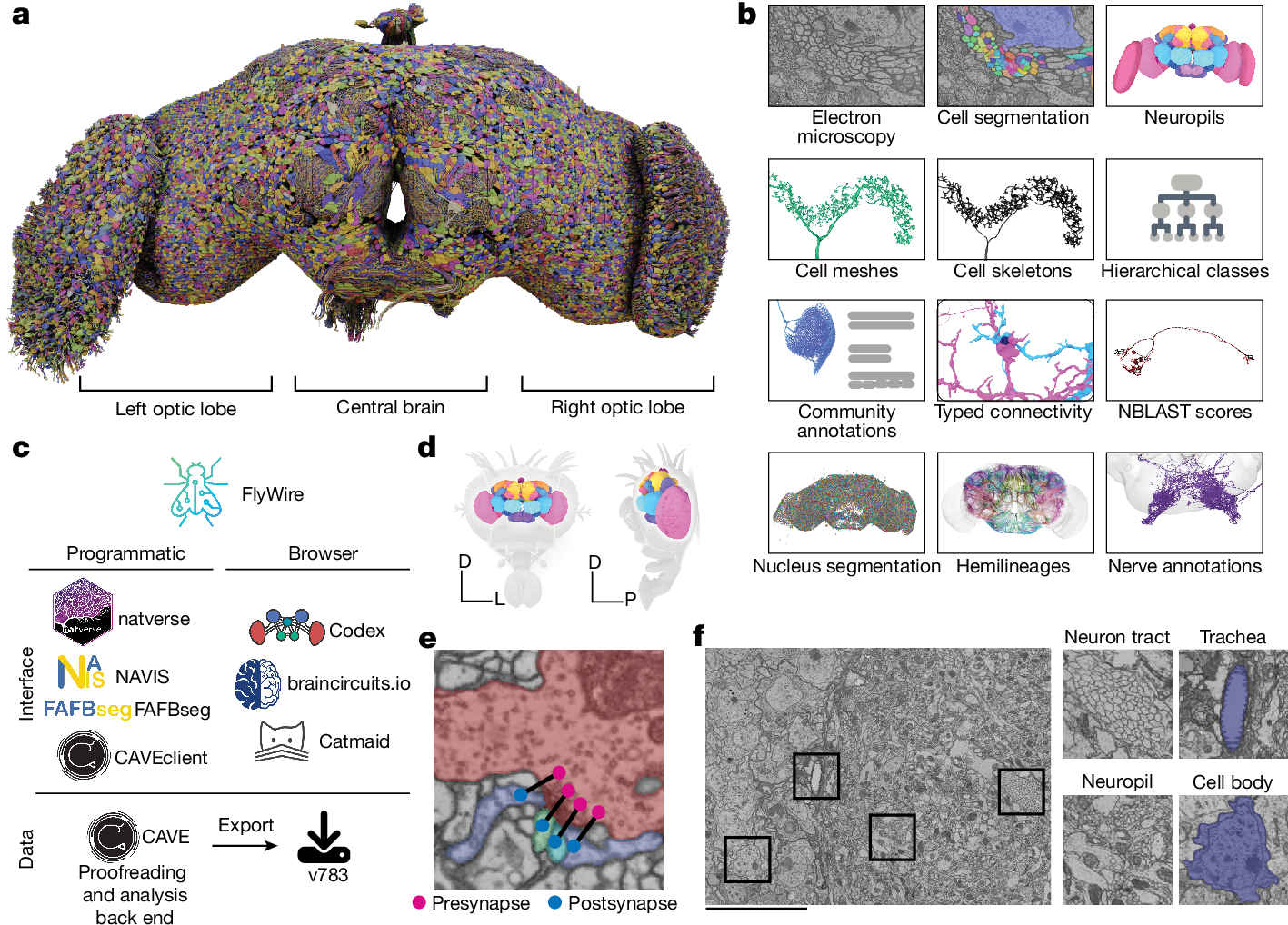
近日,FlyWire联盟等在Nature连发8篇论文,详细描述首个成年果蝇全脑连接组,并通过模拟与实验展示了它如何帮助人们形成新猜想,进而解析关键功能背后的神经环路机制。
3、The Innovation Life|医学数据计算建模:从数据采集到知识挖掘
文章从数据的采集、存在的挑战、计算建模、高性能计算到未来展望等几个方面展开进行全面的综述。比如在高性能计算方面,对并行计算、云计算、AI计算、边缘计算等几个方面都进行讨论。
最近,多所大药企和知名大学在nature子刊联合发表了关于早期药物研发数据科学的文献综述,报告了数据科学的经验指南。文章中,作者提出了数据管理和数据科学建议,以帮助实现人工智能在该领域的潜力。
该博文提出了一些关于解决基因组区间(Genomic Intervals)相关问题的方法。
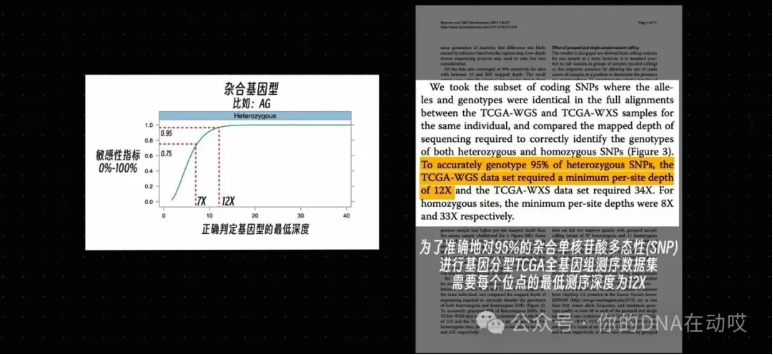
在对人类全基因组进行测序时,需要 30 ×的平均基因组覆盖率才能使局部碱基覆盖率大于 15 ×,这样才能准确检测出同源变异和异源变异。
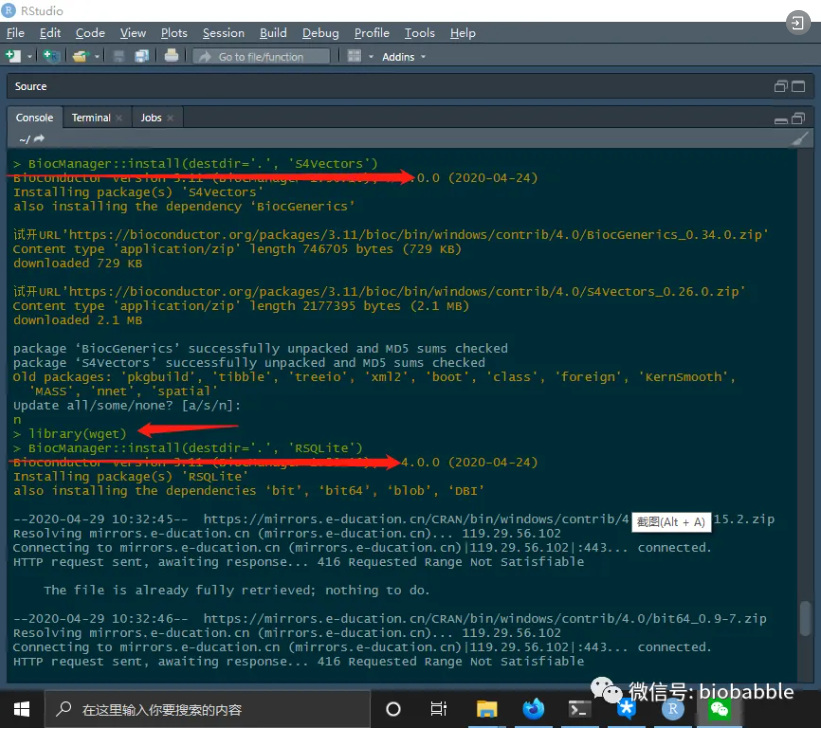
安装包有时候是个费劲的时,特别是一遍又一遍的下载安装,推荐新手朋友们使用。
8、SignalingProfiler2.0 | 网络连接多组学数据与表型特征
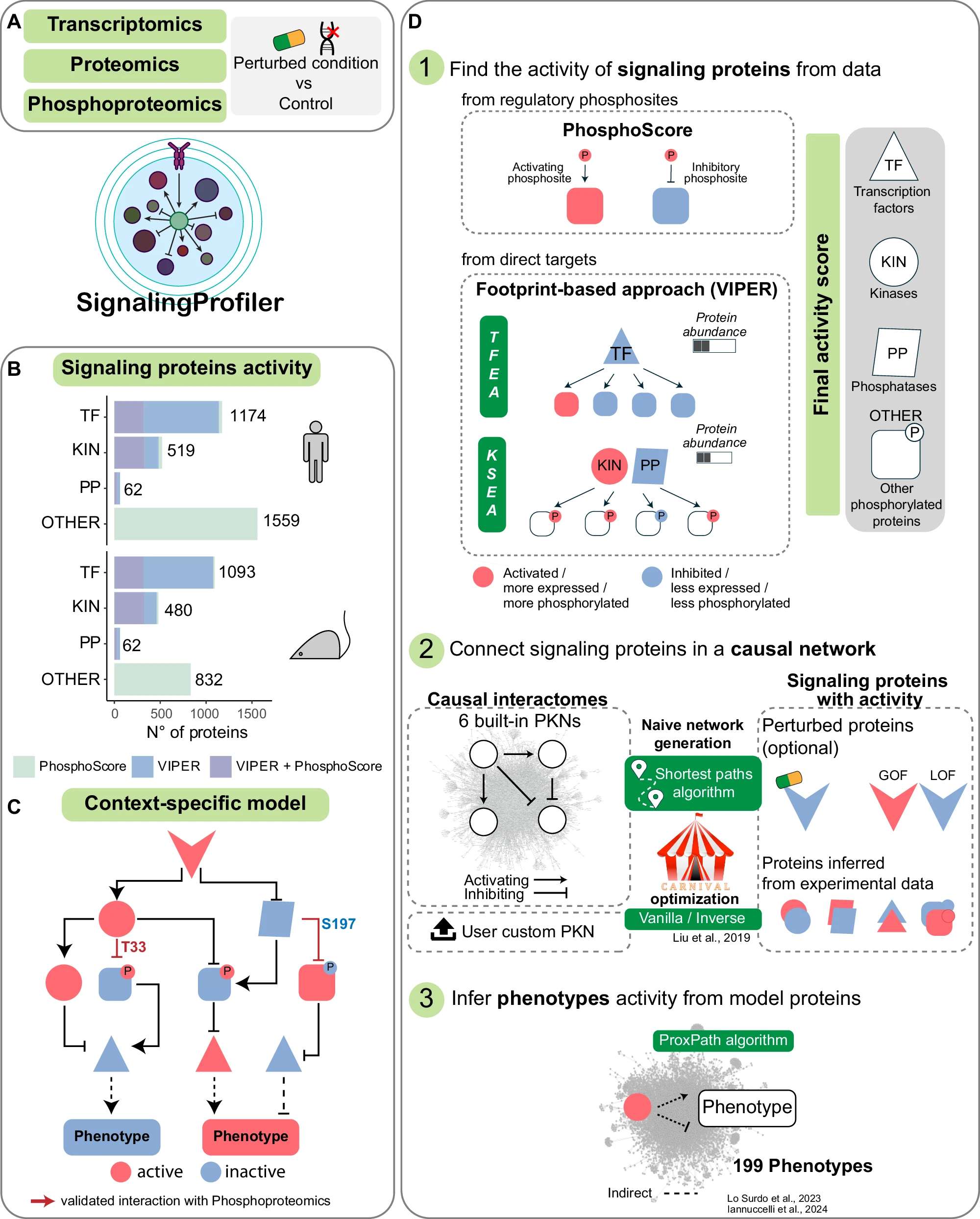
揭示细胞信号转导在受到干扰后如何重塑,对于理解疾病机制和识别潜在药物靶点至关重要。 SignalingProfiler 2.0结合蛋白基因组数据与先验知识因果网络,推导出特定情境下的信号传导网络。该工具自由访问且灵活,结合了统计学、足迹分析和图算法,加速了多组学数据的整合与解读。
9、GenomeSyn 插件 | 快速轻松展示两个基因组组装的差异情况
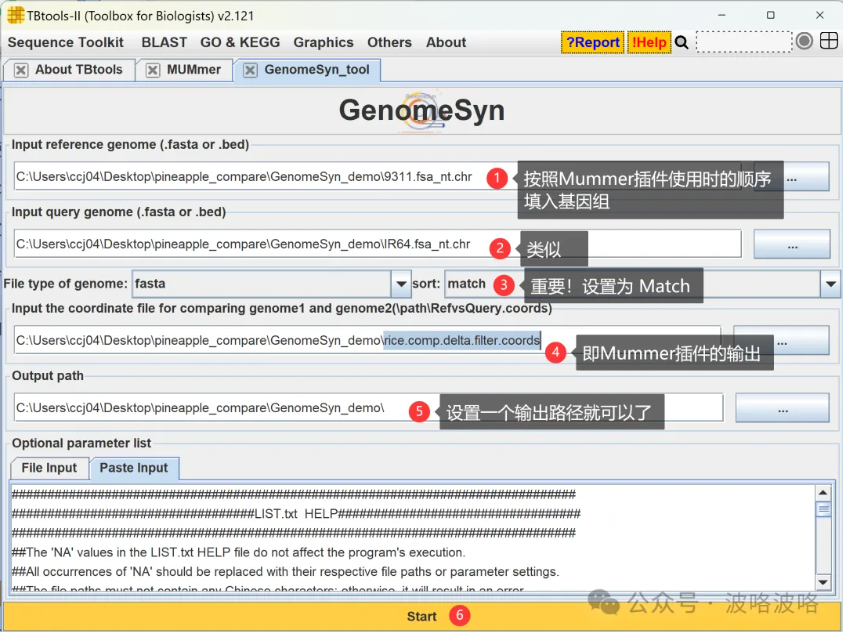
GenomeSyn是一种用于基因组同一性和结构变异分析的生物信息学可视化工具,填上Mummer 插件上输入和输出的结果,调整sort参数选项,就可以得到可视化结果。

文中列举了2024年的可投稿的生物信息学期刊及其分区,推荐给需要生信选刊的朋友们。
11、2024 年 12 个最佳 JavaScript 动画库,为您的 Web 项目提供动力

文中推荐了12 个方便有用的JavaScript 动画库,可以将绘制动态的、引人注目的动画,使Web项目更加丰富多彩。
「Openbiox 生信周刊」运维小队:
@ShixiangWang(王诗翔)@kkjtmac(阚科佳)@NiEntropy(赵启祥)@He-Kai-fly(何凯)@JnanZhang(张佳楠)@Tomcxf(陈啸枫)@wangdepin(王德品)@kongjianyang(空间阳)@donghongyu2020(董弘禹)@DrRobinLuo(罗鹏)@Wangcy-rachel - 王春阳@zoe3251 - 舒晨阳这个周刊每周日发布,同步更新在微信公众号「优雅R」(elegant-r)上。
微信搜索“优雅R”或者扫描二维码,即可订阅。
(完)
2024-10-14 23:05:44
这里记录每周值得分享的生信相关内容,周日发布。
本杂志开源(GitHub: ShixiangWang/weekly),欢迎提交 issue,投稿或推荐生信相关内容。

一个良好的评估体系,才是一个科研工作者的“乌托邦”的起点,在这里,科研工作变得更加简单和纯粹,因为大家都明白,每一份经费的分配,依据的是科研的质量和科研人员的能力和潜质,而这里的青年科学家,从踏入科学界开始,他们就知道,最重要的是要解决真正有影响力的重大问题,而不是迫于评估压力去凑齐所需的论文数目。
1、 Nat Commun|肿瘤和匹配正常样本综合突变谱MSK-IMPACT Heme可加强对血液肿瘤的临床评估
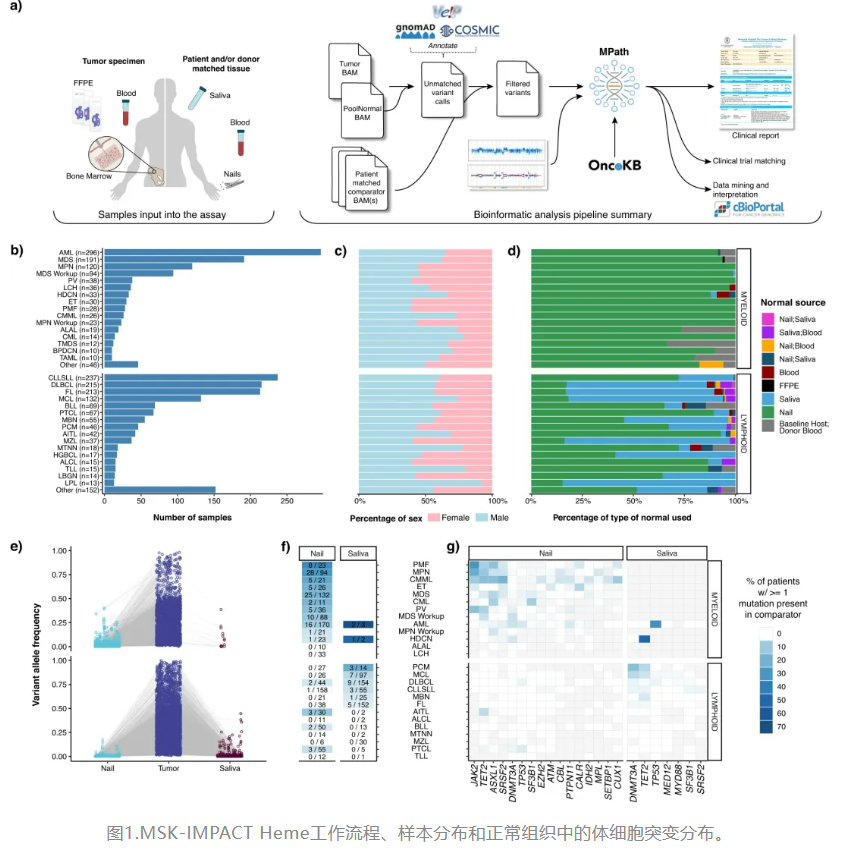
美国纪念斯隆-凯特琳癌症中心(MSKCC)研究团队通过展示MSK-IMPACT Heme的开发和临床经验分享了应对血液肿瘤体细胞突变评估的独特挑战的经验。MSK-IMPACT Heme是针对血液肿瘤可操作靶点的综合突变谱,是通过基于杂交捕获和高覆盖率的NGS分析平台获得的肿瘤和匹配正常DNA获得体细胞突变的综合数据集。该研究报告了大规模的前瞻性临床测序工作的经验,有助于指导血液肿瘤患者的诊断、预后、治疗选择和未来监测。研究表明,肿瘤和匹配正常样本的大规模测序在血液肿瘤中是可行的,包括造血干细胞移植后的患者和患有多种并发恶性肿瘤的患者。这一流程的周转时间是可变的,在该队列中通常为2-3周。 - 文章链接:https://www.nature.com/articles/s41467-023-42585-9
2、Genome Medicine | 对两例高危前列腺癌的演化分析揭示个性化医疗前景
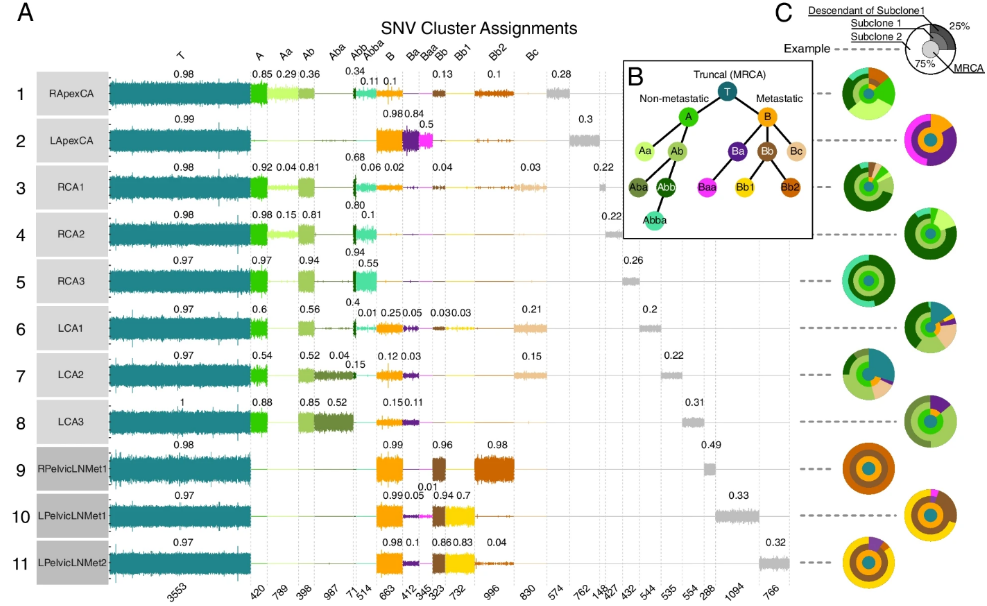
文章采用全新的处理技术,对两名接受根治性前列腺切除术的高危PrCa患者(GP5和GP12)进行全基因组数据以及三维解剖和组织形态学分析。并利用进化重建工具和空间进化模型分析了22个全基因组测序位点(16个原发癌灶和6个淋巴结转移)。概率模型用于追踪原发肿瘤和转移的空间和时间起源,绘制其遗传驱动因素,并区分转移性和非转移性亚克隆。 - 文章链接:https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-023-01242-y
3、Nat Commun |长读长转录组数据的分析工具包TAGET

北京大学数学科学学院的席瑞斌团队和哈尔滨医科大学附属肿瘤医院苗素生团队在Nature Communications上发表了题为“TAGET: A toolkit for analyzing full-length transcripts from long-read sequencing”的文章。研究团队提出了一个名为TAGET(Toolkit for Analyzing full-length GEne Transcripts)的综合性计算工具包,用来分析Iso-seq全长转录组数据,包括转录本的映射和注释、基因融合的检测、差异表达分析以及差异同源异构体使用(DIU)分析。通过实验验证和与RNA-seq数据的比较,研究团队发现TAGET比其它工具更准确地检测新剪切位点、同源异构体和基因融合。这项研究进一步完善了三代测序Iso-seq数据的分析流程,帮助研究人员更方便、更准确地进行全长转录组分析。
4、博文资讯 | 揭秘环形RNA的“生老病死”与“独门秘诀”|上海市科学技术奖
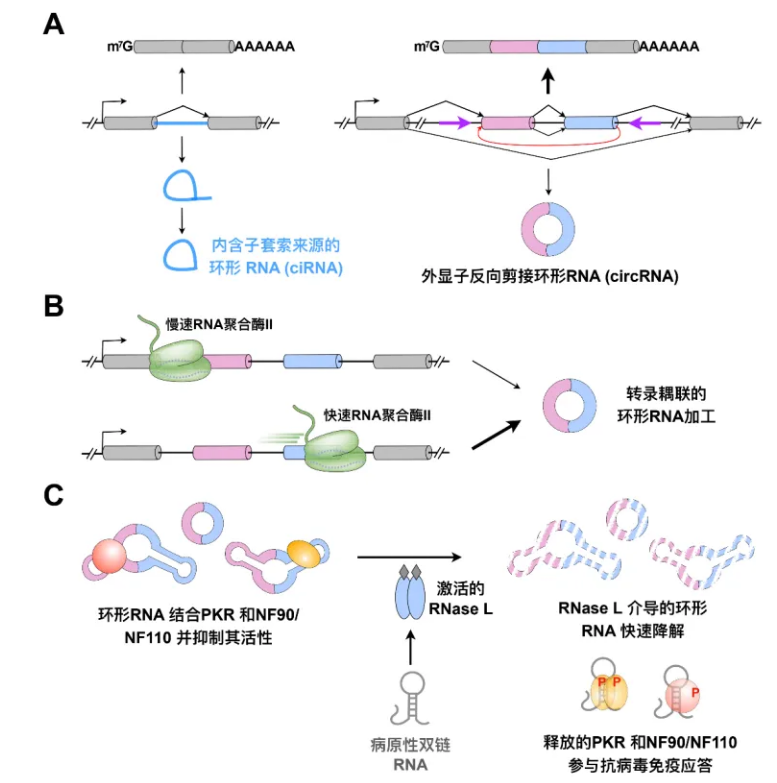
陈玲玲研究员围绕着“环形RNA生成和功能机制的研究”的项目进行研究,通过系统阐明环形RNA生成的反式作用因子、折叠和降解的规律,创新性地将环形RNA的生成与作用机制相耦联,揭示其全新功能并与自身免疫病密切关联。这一项目荣获2022年的上海市自然科学奖一等奖。

NIH正在建立人类健康与疾病的多组学联盟(Multi-Omics for Health and Disease Consortium),首年拨款约1100万美元,该联盟旨在促进人类健康研究多组学数据的生成和分析。NIH将在五年内为多组学联盟提供共约5030万美元的资助。资金由美国国家人类基因组研究所、美国国家癌症研究所(NCI)以及美国国家环境健康科学研究所(NIEHS)共同提供。
6、R语言相关性热图汇总之LinkET篇(下):网络相关性热图跟方形相关性热图
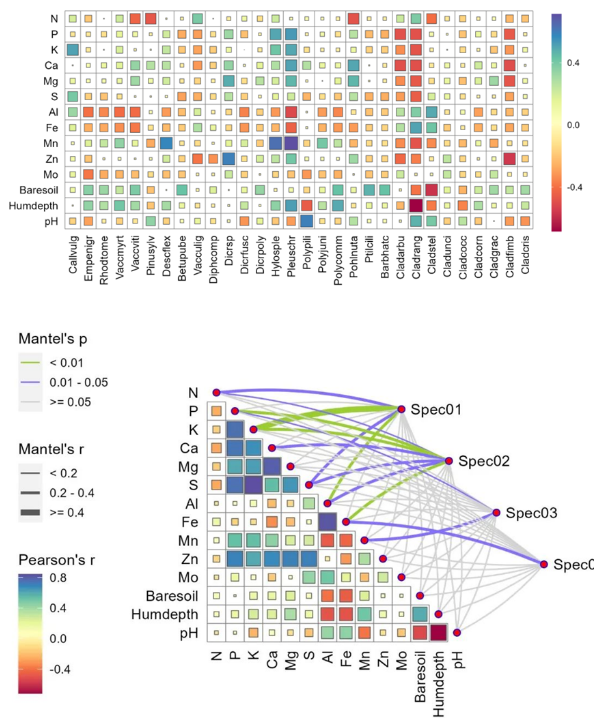
本篇为LinkET包来绘制相关性热图的下篇,包括方形相关性热图及网络相关性热图,上篇见R语言相关性热图汇总之LinkET篇(上)。 主要介绍了方形相关性热图和网络相关性热图的用途和代码实现过程。
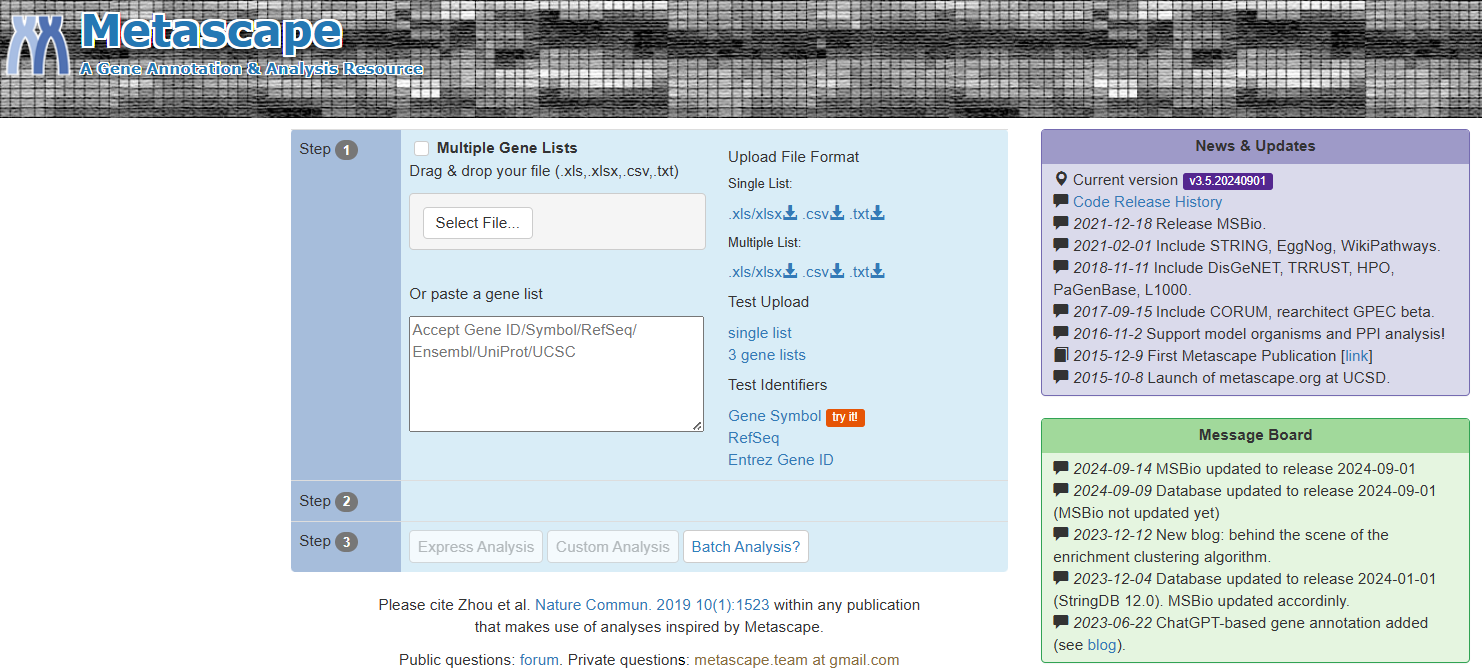
Metascape的使用非常简单,只要将基因列表贴入提交,然后点击Express Analysis按钮即可。Metascape能自动识别常用的各种基因或蛋白质的标识符。分析完成后网页会引导用户打开一份分析报告。分析报告模仿科研论文的格式来展现分析结果,图文并茂,对生物学者极其友好。报告中详细阐述了分析方法和图形的意义,而且图形都含可以发表的高清晰文件格式。报告还提供了格式化好的Excel文件,许多文章直接使用它做supplementary table。自动生成的PowerPoint文件方便学者们交流结果。所有的数据和图标文件都可以通过一个Zip文件包下载保存。蛋白质网络文件格式还支持用第三方软件比如Cytoscape进行更加深入的分析。用户熟悉后也可以使用Custom Analysis按钮对更多的分析功能和参数予以调节。

根据需求一步步设计品牌Logo,感兴趣可以尝试
9、Rfast
一组用于数据分析的快速(实用)函数。列和行方向上的平均值、中位数、方差、最小值、最大值,以及许多t检验、F检验和G平方检验,还有多种回归分析(正态、逻辑斯蒂、泊松),这些都是众多快速函数中的一部分。

提供中文文档快速入门页面开发,简单易用,适配性强。 - Github: https://github.com/zh-lx/code-inspector

为疲惫的数据科学家准备的SQL入门指南。
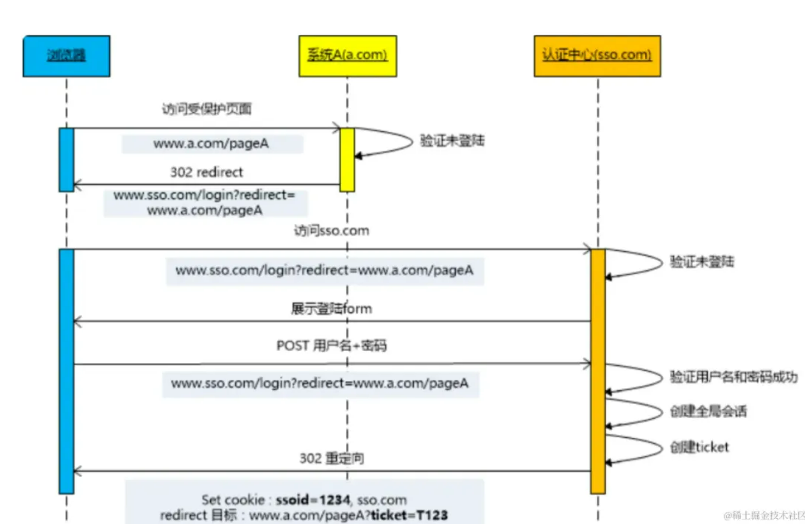
单点登录,首先可以想到传统登录,通过登录页面根据用户名查询用户信息,判断密码是否正确,正确则将用户信息写到session,访问的时候通过从session中获取用户信息,判断是否已登录,登录则允许访问。
「Openbiox 生信周刊」运维小队:
@ShixiangWang(王诗翔)@kkjtmac(阚科佳)@NiEntropy(赵启祥)@He-Kai-fly(何凯)@JnanZhang(张佳楠)@Tomcxf(陈啸枫)@wangdepin(王德品)@kongjianyang(空间阳)@donghongyu2020(董弘禹)@DrRobinLuo(罗鹏)这个周刊每周日发布,同步更新在微信公众号「优雅R」(elegant-r)上。
微信搜索“优雅R”或者扫描二维码,即可订阅。
(完)